
1. What is the Allele-Specific DNA Methylation Database?
DNA methylation plays a crucial role in most organisms. Bisulfite sequencing (BS-Seq) is an approach that can be used to obtain quantitative cytosine methylation levels at a genome-wide scale and single-base resolution. Besides, parental alleles in haploids might exhibit different methylation patterns, which can lead to different phenotypes and even different therapeutic and drug responses to diseases. A comprehensive database of high-throughput BS-Seq data and allele-specific DNA methylation (ASM) results from multiple species was not available prior to this study.
Here, we constructed The Allele-Specific DNA Methylation Databases (ASMdb), aiming to provide a comprehensive resource and a web tool for showing the DNA methylation level and differential DNA methylation in diverse organisms, including 47 species, such as Homo sapiens, Mus musculus, Arabidopsis thaliana, and Oryza Sativa, with 5998 GEO samples (4400 BS-Seq data and 1598 RNA-Seq data).
2. The pipeline of database construction
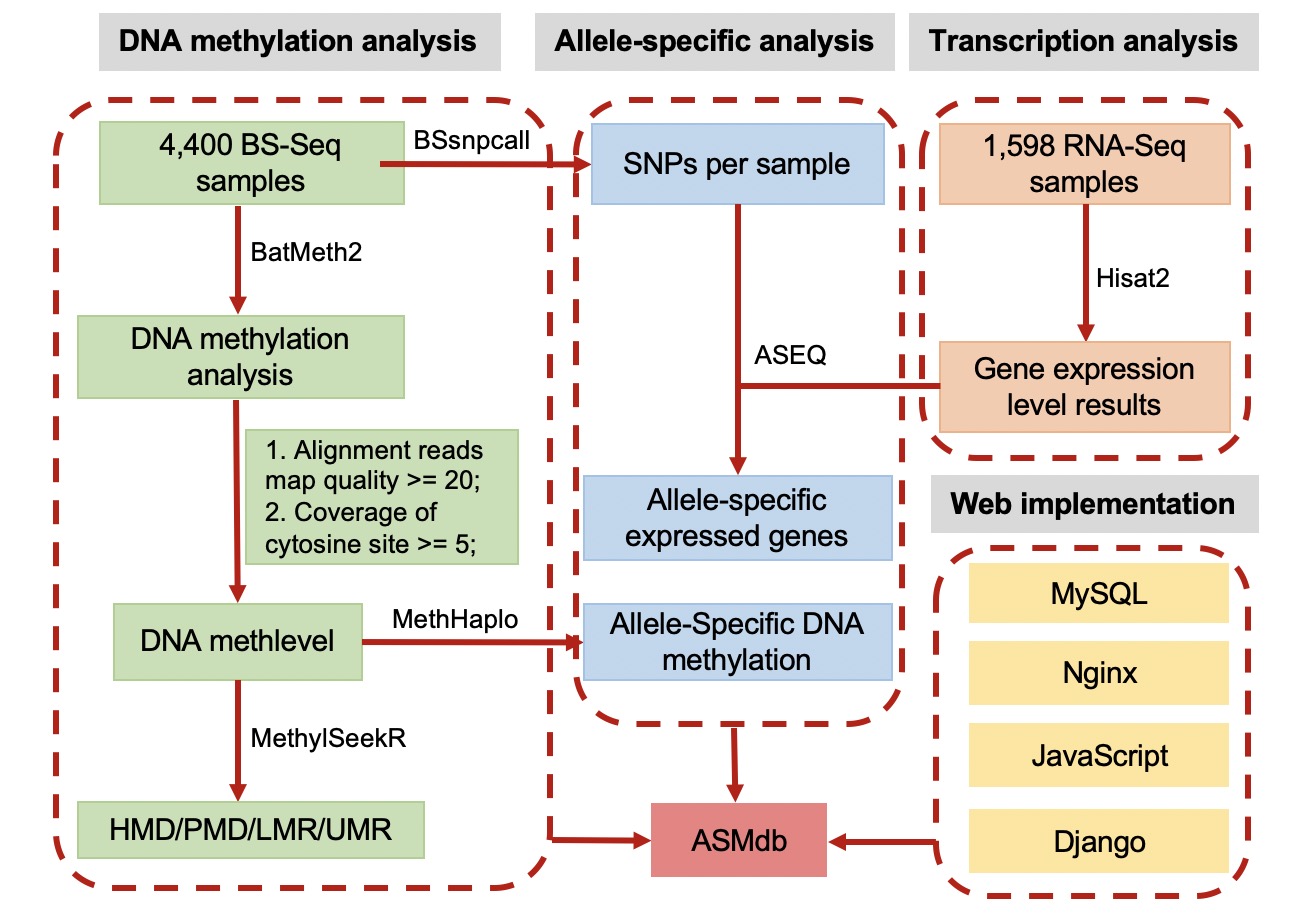
3. Processing of methylation data
4. SNP calling with BS-Seq data
5. Identification of ASM
6. Identification of allele-specific expression genes (ASEGs)
7. Identification of PMD/HMD/LMR/UMR
#MethylSeekR
install.packages("BiocManager")
BiocManager::install("MethylSeekR")
library(MethylSeekR)
library(BSgenome)
library(parallel)
library(rtracklayer)
BiocManager::install("BSgenome")
library(BSgenome)
#available.genomes()
BiocManager::install("BSgenome.Osativa.MSU.MSU7")
library("BSgenome.Osativa.MSU.MSU7")
build <- get("BSgenome.Osativa.MSU.MSU7")
#also can bulid by ourself..
#library(BSgenome.Astyanax.mexicanus)
#build <- get("BSgenome.Astyanax.mexicanus")
Args <- commandArgs()
faifile<-Args[6] ##faifile
pref<-Args[7] ## pref
cpgisland<-Args[8] ##cpgislad
trainchr<-Args[9] ##train chr
fai <- read.delim(faifile, header=F)
chromosome_lengths <- fai$V2
names(chromosome_lengths) <- fai$V1
d <- readMethylome(paste(pref, "_loci.CG.txt.methylseekr", sep=""), chromosome_lengths)
snps.gr <- readSNPTable(paste(pref, ".snp.methylseekr", sep="") , chromosome_lengths)
## removr snp
meth.gr <- removeSNPs(d, snps.gr)
sample = pref
num.cores=4
pdf(paste0(sample, ".pdf"), width=14)
PMDsegments.gr <- segmentPMDs(m=meth.gr, chr.sel=trainchr, seqLengths=chromosome_lengths, num.cores=num.cores)
##
CpGisland <- import(cpgisland, format="bed")
write.csv(calculateFDRs(meth.gr, CpGisland, PMDs=PMDsegments.gr, num.cores=num.cores)$FDRs, paste0(sample,".PMD.stats"))
write.csv(calculateFDRs(meth.gr, CpGisland, num.cores=num.cores)$FDRs, paste0(sample,".stats"))
### UMR LMR
PMD.UMRLMR <- segmentUMRsLMRs(m=meth.gr, meth.cutoff=0.5, nCpG.cutoff=5, num.cores=num.cores, myGenomeSeq=build, seqLengths=chromosome_lengths, PMDs=PMDsegments.gr)
# Repeat without filtering PMDs
UMRLMR <- segmentUMRsLMRs(m=meth.gr, meth.cutoff=0.5, nCpG.cutoff=5, num.cores=num.cores, myGenomeSeq=build, seqLengths=chromosome_lengths)
dev.off()
# Export to bed/bigbed
tmp <- list("PMD"=PMDsegments.gr[PMDsegments.gr$type=="PMD"],
"UMR"=UMRLMR[UMRLMR$type=="UMR"],
"LMR"=UMRLMR[UMRLMR$type=="LMR"],
"PMD.UMR"=PMD.UMRLMR[PMD.UMRLMR$type=="UMR"],
"PMD.LMR"=PMD.UMRLMR[PMD.UMRLMR$type=="LMR"])
chrom.sizes <- data.frame(seqlevels(build), seqlengths(build))
write.table(chrom.sizes, paste0(sample, ".chrom.sizes"), quote=FALSE, sep="\t", row.names=FALSE, col.names=FALSE)
for (j in names(tmp)) {
bed.file.name <- paste(sample, j, "bed", sep=".")
export(tmp[[j]], bed.file.name, format="bed")
system(paste0("cut -f1,2,3 ", bed.file.name, " > tmp; /public/home/qwzhou/software/EMBOSS-6.6.0/bin/bedToBigBed tmp ", sample, ".chrom.sizes ", gsub(".bed$", ".bb", bed.file.name), "; rm tmp"))
}
8. Genome List
| Species | Genome | Genome FTP | Annotation FTP |
|---|---|---|---|
| Homo sapiens | GRCh38.p12 | ftp://ftp.ensembl.org/pub/release-96/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz | ftp://ftp.ensembl.org/pub/release-96/gff3/homo_sapiens/Homo_sapiens.GRCh38.96.chr.gff3.gz |
| Mus musculus | GRCm38.p6 | ftp://ftp.ensembl.org/pub/release-96/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.toplevel.fa.gz | ftp://ftp.ensembl.org/pub/release-96/gff3/mus_musculus/Mus_musculus.GRCm38.96.chr.gff3.gz |
| Arabidopsis thaliana | TAIR10 | ftp://ftp.arabidopsis.org/home/tair/Sequences/whole_chromosomes/ | ftp://ftp.arabidopsis.org/home/tair/Sequences/whole_chromosomes/ |
| Astyanax mexicanus | Astyanax mexicanus 2.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/372/685/GCF_000372685.2_Astyanax_mexicanus-2.0/GCF_000372685.2_Astyanax_mexicanus-2.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/372/685/GCF_000372685.2_Astyanax_mexicanus-2.0/GCF_000372685.2_Astyanax_mexicanus-2.0_genomic.gff.gz |
| Bos indicus x Bos taurus | UMD3.1 | http://128.206.12.216/drupal/sites/bovinegenome.org/files/data/umd3.1/UMD3.1_chromosomes.fa.gz | http://128.206.12.216/drupal/sites/bovinegenome.org/files/data/umd3.1/Ensembl79_UMD3.1_genes.gff3.gz |
| Bos taurus | UMD3.1 | http://128.206.12.216/drupal/sites/bovinegenome.org/files/data/umd3.1/UMD3.1_chromosomes.fa.gz | http://128.206.12.216/drupal/sites/bovinegenome.org/files/data/umd3.1/Ensembl79_UMD3.1_genes.gff3.gz |
| Camellia sinensis var. assamica | Tea_Tree_2017 | http://www.plantkingdomgdb.com/tea_tree/data/assembly/Teatree_Assembly.fas | http://www.plantkingdomgdb.com/tea_tree/data/gff3/Teatree.gff3 |
| Camponotus floridanus | Cflo_v7.5 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/227/725/GCF_003227725.1_Cflo_v7.5/GCF_003227725.1_Cflo_v7.5_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/227/725/GCF_003227725.1_Cflo_v7.5/GCF_003227725.1_Cflo_v7.5_genomic.gff.gz |
| Canis lupus familiaris | CanFam3.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/285/GCF_000002285.3_CanFam3.1/GCF_000002285.3_CanFam3.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/285/GCF_000002285.3_CanFam3.1/GCF_000002285.3_CanFam3.1_genomic.gff.gz |
| Ceratina calcarata | C. calcarata_v1.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/652/005/GCF_001652005.1_ASM165200v1/GCF_001652005.1_ASM165200v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/652/005/GCF_001652005.1_ASM165200v1/GCF_001652005.1_ASM165200v1_genomic.gff.gz |
| Citrus sinensis | Citrus sinensis (version2) | http://citrus.hzau.edu.cn/orange/download/csi.chromosome.fa.tar.gz | http://citrus.hzau.edu.cn/orange/download/csi.gene.models.gff3.tar.gz |
| Oryza sativa | MSU7.0 | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.con | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.gff3 |
| Oryza sativa Japonica Group | MSU7.0 | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.con | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.gff3 |
| Oryza sativa Japonica Group x Oryza sativa Indica Group | MSU7.0 | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.con | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.gff3 |
| Oryza sativa Indica Group | MSU7.0 | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.con | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.gff3 |
| Oryza sativa Indica Group x Oryza sativa Japonica Group | MSU7.0 | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.con | http://rice.plantbiology.msu.edu/pub/data/EukaryoticProjects/osativa/annotationdbs/pseudomolecules/version7.0/all.dir/all.gff3 |
| Cordyceps militaris | CM01 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/225/605/GCF_000225605.1_CmilitarisCM01_v01/GCF_000225605.1_CmilitarisCM01_v01_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/225/605/GCF_000225605.1_CmilitarisCM01_v01/GCF_000225605.1_CmilitarisCM01_v01_genomic.gff.gz |
| Cynoglossus semilaevis | Cse_v1.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/523/025/GCF_000523025.1_Cse_v1.0/GCF_000523025.1_Cse_v1.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/523/025/GCF_000523025.1_Cse_v1.0/GCF_000523025.1_Cse_v1.0_genomic.gff.gz |
| Danio rerio | GRCz11 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/035/GCF_000002035.6_GRCz11/GCF_000002035.6_GRCz11_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/035/GCF_000002035.6_GRCz11/GCF_000002035.6_GRCz11_genomic.gff.gz |
| Daphnia magna | dmagna-v2.4-20100422 | http://wfleabase.org/genome/Daphnia_magna/openaccess/genome/dmagna-v2.4-20100422-assembly.fna.gz | http://wfleabase.org/genome/Daphnia_magna/openaccess/genes/Genes/earlyaccess/dmagset7finloc9c.puban.gff.gz |
| Dorcoceras hygrometricum | Boea_hygrometrica.v1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/001/598/015/GCA_001598015.1_Boea_hygrometrica.v1/GCA_001598015.1_Boea_hygrometrica.v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/001/598/015/GCA_001598015.1_Boea_hygrometrica.v1/GCA_001598015.1_Boea_hygrometrica.v1_genomic.gff.gz |
| Zea mays | B73_v4 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/005/GCF_000005005.2_B73_RefGen_v4/GCF_000005005.2_B73_RefGen_v4_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/005/GCF_000005005.2_B73_RefGen_v4/GCF_000005005.2_B73_RefGen_v4_genomic.gff.gz |
| Trichodesmium | IMS101 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.gff.gz |
| Trichodesmium erythraeum 21-75 | 21-57 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/963/755/GCF_000963755.1_ASM96375v2/GCF_000963755.1_ASM96375v2_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/963/755/GCF_000963755.1_ASM96375v2/GCF_000963755.1_ASM96375v2_genomic.gff.gz |
| Trichodesmium erythraeum IMS101 | IMS101 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.gff.gz |
| Trichodesmium thiebautii VI-1 | IMS101 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/014/265/GCF_000014265.1_ASM1426v1/GCF_000014265.1_ASM1426v1_genomic.gff.gz |
| Tribolium castaneum | Tcas_v5.2 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/335/GCF_000002335.3_Tcas5.2/GCF_000002335.3_Tcas5.2_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/335/GCF_000002335.3_Tcas5.2/GCF_000002335.3_Tcas5.2_genomic.gff.gz |
| Sus scrofa | Sscrofa11.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/003/025/GCF_000003025.6_Sscrofa11.1/GCF_000003025.6_Sscrofa11.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/003/025/GCF_000003025.6_Sscrofa11.1/GCF_000003025.6_Sscrofa11.1_genomic.gff.gz |
| Sorghum bicolor | Sbi1.4 | ftp://ftp.jgi-psf.org/pub/JGI_data/Sorghum_bicolor/v1.0/Sbi/assembly/Sbi1/sbi1.fasta.gz | ftp://ftp.jgi-psf.org/pub/JGI_data/Sorghum_bicolor/v1.0/Sbi/annotation/Sbi1.4/Sbi1.4.gff3.gz |
| Rattus norvegicus | Rnor6.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/895/GCF_000001895.5_Rnor_6.0/GCF_000001895.5_Rnor_6.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/895/GCF_000001895.5_Rnor_6.0/GCF_000001895.5_Rnor_6.0_genomic.gff.gz |
| Procambarus virginalis | Pvir0.4 | http://marmorkrebs.dkfz.de/downloads/genome/pvirGEN-0.4/Pvir04.fa.gz | http://marmorkrebs.dkfz.de/downloads/genome/pvirGEN-0.4/Pvir04.annotation.gff.gz |
| Procambarus fallax | Pvir0.4 | http://marmorkrebs.dkfz.de/downloads/genome/pvirGEN-0.4/Pvir04.fa.gz | http://marmorkrebs.dkfz.de/downloads/genome/pvirGEN-0.4/Pvir04.annotation.gff.gz |
| Physcomitrella patens | PpatensV3.3 | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Ppatens | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Ppatens |
| Pan troglodytes | PanTro5/pan_tro3.0 | ftp://ftp.ensembl.org/pub/release-96/fasta/pan_troglodytes/dna/Pan_troglodytes.Pan_tro_3.0.dna.toplevel.fa.gz | ftp://ftp.ensembl.org/pub/release-96/gff3/pan_troglodytes/Pan_troglodytes.Pan_tro_3.0.96.chr.gff3.gz |
| Neurospora crassa | NC12 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/182/925/GCF_000182925.2_NC12/GCF_000182925.2_NC12_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/182/925/GCF_000182925.2_NC12/GCF_000182925.2_NC12_genomic.gff.gz |
| Nasonia vitripennis | Nvit2.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.gff.gz |
| Nasonia vitripennis x Nasonia giraulti | Nvit2.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.gff.gz |
| Erythranthe guttata | Mguttata2.0 | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Mguttatus | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Mguttatus |
| Erythranthe lutea x Erythranthe guttata | Mguttata2.0 | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Mguttatus | https://genome.jgi.doe.gov/portal/pages/dynamicOrganismDownload.jsf?organism=Mguttatus |
| Erythranthe lutea | Mimulus luteus v1.0 | https://genomevolution.org/coge/GenomeInfo.pl?gid=22585 | https://genomevolution.org/coge/GenomeInfo.pl?gid=22585 |
| Glycine max | Glyma2.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/004/515/GCF_000004515.5_Glycine_max_v2.1/GCF_000004515.5_Glycine_max_v2.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/004/515/GCF_000004515.5_Glycine_max_v2.1/GCF_000004515.5_Glycine_max_v2.1_genomic.gff.gz |
| Gorilla gorilla | gorGor4 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/151/905/GCF_000151905.2_gorGor4/GCF_000151905.2_gorGor4_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/151/905/GCF_000151905.2_gorGor4/GCF_000151905.2_gorGor4_genomic.gff.gz |
| Harpegnathos saltator | Hsal_v8.5 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/227/715/GCF_003227715.1_Hsal_v8.5/GCF_003227715.1_Hsal_v8.5_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/227/715/GCF_003227715.1_Hsal_v8.5/GCF_003227715.1_Hsal_v8.5_genomic.gff.gz |
| Macaca fuscata | Mmul_10 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/339/765/GCF_003339765.1_Mmul_10/GCF_003339765.1_Mmul_10_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/339/765/GCF_003339765.1_Mmul_10/GCF_003339765.1_Mmul_10_genomic.gff.gz |
| Macaca mulatta | Mmul_10 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/339/765/GCF_003339765.1_Mmul_10/GCF_003339765.1_Mmul_10_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/339/765/GCF_003339765.1_Mmul_10/GCF_003339765.1_Mmul_10_genomic.gff.gz |
| Mimulus peregrinus | M. guttatus concatenate M. luteus. | M. guttatus concatenate M. luteus. | M. guttatus concatenate M. luteus. |
| Nasonia giraulti | Nvit2.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/325/GCF_000002325.3_Nvit_2.1/GCF_000002325.3_Nvit_2.1_genomic.gff.gz |
| Nomascus leucogenys | Nleu_3.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/146/795/GCF_000146795.2_Nleu_3.0/GCF_000146795.2_Nleu_3.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/146/795/GCF_000146795.2_Nleu_3.0/GCF_000146795.2_Nleu_3.0_genomic.gff.gz |
| Xenopus laevis | Xenopus_laevis_v2 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/663/975/GCF_001663975.1_Xenopus_laevis_v2/GCF_001663975.1_Xenopus_laevis_v2_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/663/975/GCF_001663975.1_Xenopus_laevis_v2/GCF_001663975.1_Xenopus_laevis_v2_genomic.gff.gz |
| Xenopus tropicalis x Xenopus laevis | Xenopus_laevis_v2 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/663/975/GCF_001663975.1_Xenopus_laevis_v2/GCF_001663975.1_Xenopus_laevis_v2_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/001/663/975/GCF_001663975.1_Xenopus_laevis_v2/GCF_001663975.1_Xenopus_laevis_v2_genomic.gff.gz |
| Plectus sambesii | P_sam | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/plectus_sambesii/PRJNA390260/plectus_sambesii.PRJNA390260.WBPS13.genomic.fa.gz | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/plectus_sambesii/PRJNA390260/plectus_sambesii.PRJNA390260.WBPS13.annotations.gff3.gz |
| Trichuris muris | T_muris | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/trichuris_muris/PRJEB126/trichuris_muris.PRJEB126.WBPS13.genomic.fa.gz | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/trichuris_muris/PRJEB126/trichuris_muris.PRJEB126.WBPS13.annotations.gff3.gz |
| Trichinella spiralis | T_spir | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/trichinella_spiralis/PRJNA257433/trichinella_spiralis.PRJNA257433.WBPS13.genomic.fa.gz | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/trichinella_spiralis/PRJNA257433/trichinella_spiralis.PRJNA257433.WBPS13.annotations.gff3.gz |
| Romanomermis culicivorax | R_culici | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/romanomermis_culicivorax/PRJEB1358/romanomermis_culicivorax.PRJEB1358.WBPS13.genomic.fa.gz | ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS13/species/romanomermis_culicivorax/PRJEB1358/romanomermis_culicivorax.PRJEB1358.WBPS13.annotations.gff3.gz |
| Drosophila melanogaster | BDGP6.22 | ftp://ftp.ensembl.org/pub/release-96/fasta/drosophila_melanogaster/dna/Drosophila_melanogaster.BDGP6.22.dna.toplevel.fa.gz | ftp://ftp.ensembl.org/pub/release-96/gff3/drosophila_melanogaster/Drosophila_melanogaster.BDGP6.22.96.chr.gff3.gz |
| Solanum lycopersicum | SL3.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/188/115/GCF_000188115.4_SL3.0/GCF_000188115.4_SL3.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/188/115/GCF_000188115.4_SL3.0/GCF_000188115.4_SL3.0_genomic.gff.gz |
| Apis mellifera | HAv3.1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/254/395/GCF_003254395.2_Amel_HAv3.1/GCF_003254395.2_Amel_HAv3.1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/254/395/GCF_003254395.2_Amel_HAv3.1/GCF_003254395.2_Amel_HAv3.1_genomic.gff.gz |
| Pyrus x bretschneideri | Pbr_v1.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/315/295/GCF_000315295.1_Pbr_v1.0/GCF_000315295.1_Pbr_v1.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/315/295/GCF_000315295.1_Pbr_v1.0/GCF_000315295.1_Pbr_v1.0_genomic.gff.gz |
| Chlamydomonas reinhardtii | Crein_v3.0 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/595/GCF_000002595.1_v3.0/GCF_000002595.1_v3.0_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/595/GCF_000002595.1_v3.0/GCF_000002595.1_v3.0_genomic.gff.gz |
| Marchantia polymorpha | Mpoly_v1 | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/003/032/435/GCA_003032435.1_Marchanta_polymorpha_v1/GCA_003032435.1_Marchanta_polymorpha_v1_genomic.fna.gz | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/003/032/435/GCA_003032435.1_Marchanta_polymorpha_v1/GCA_003032435.1_Marchanta_polymorpha_v1_genomic.gff.gz |
9. Jbrowse Usage